


Infection with the human immunodeficiency virus (HIV) involves the integration of the viral genome into host cells, which can lead to proviral latency in certain cellular populations, such as resting memory CD4+ T-cells. Despite effective antiretroviral therapy (ART), integrated viral genomes persist in these cellular reservoirs and can cause viral rebound upon treatment interruption. Since not all individuals can access or adhere to ART indefinitely, there is an urgent need for therapeutic interventions that suppress or eliminate these cellular HIV reservoirs. However, the complete characterization of the HIV reservoir in individuals receiving effective ART remains elusive, due in part to the scarcity of replication-competent proviruses in these patients’ samples, estimated at 1 in 100,000 CD4+ T-cells. Current sequencing strategies face limitations in amplifying these components from individual DNA molecules and are prone to artifact generation during amplification and PCR. To address these challenges, Sun et al developed SIP-seq, a droplet microfluidics-based assay enabling simultaneous integration site and provirus sequencing. SIP-seq facilitates comprehensive profiling of HIV proviruses and adjacent host integration sites in individuals receiving effective ART, shedding light on the genetics of the latent HIV reservoir.

The goal of the SIP-seq assay is to recover and sequence all DNA fragments containing proviral genomes from a patient sample. By compartmentalizing the genomic fragments within microdroplets, SIP-seq ensures the selective amplification and sequencing of each provirus separately. This individual encapsulation is essential for obtaining single provirus data, allowing for precise analysis of the HIV genome and adjacent host junctions. Compared to conventional dilution methods, the chance of having multiple viruses in a single droplet is exceptionally low, with a doublet rate below 0.0001%, compared to approximately 4.5% using conventional methods. Using a droplet generator, human genomic DNA fragments were coencapsulated with the reagents for multiple displacement amplification (MDA) and incubated at 30°C to enable whole genome amplification. These droplets were then merged with droplets containing TaqMan PCR reagents, which will tag the droplets containing proviruses for sequencing. To increase the specificity for full-length proviruses, the TaqMan assays were designed to target two conserved regions of HIV spaced >5kbp apart (HIV pol and env). Full-length proviruses would thus generate a two-color fluorescence signal, whereas partial HIV genomes would result in single positive droplets. Droplets that did not contain HIV genomes would not generate a fluorescence signal. After thermocycling, the droplets were injected into a droplet sorter for fluorescence-activated droplet sorting (FADS), which enables high-throughput selection of rare positive droplets.
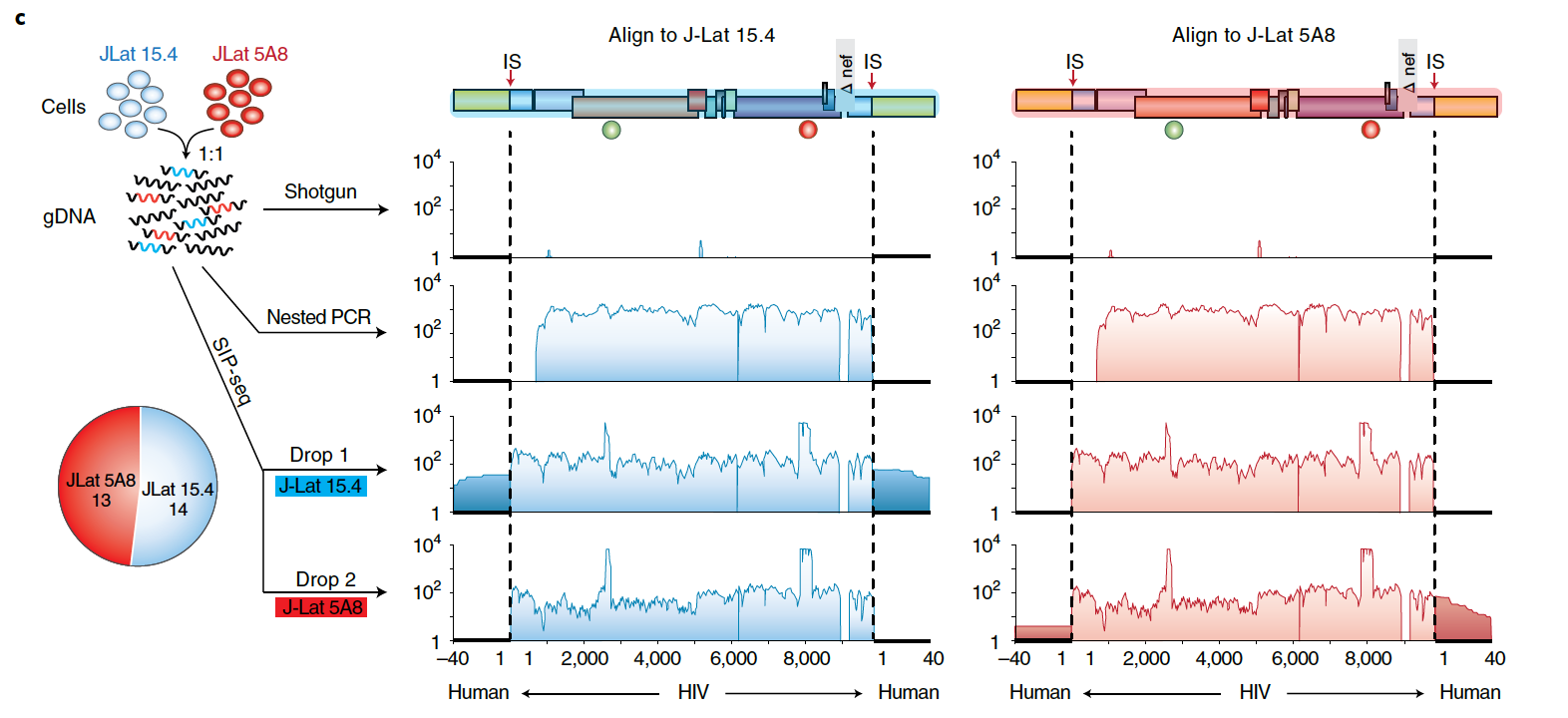
DNA from each sorted droplet was then processed for sequencing. As the small reaction volume within droplets permits the sequencing of picograms of DNA, whole genome amplification products can be sequenced directly without need for additional multi-primer amplification. SIP-seq thus yields large, gapless contiguous assemblies that contain both the entire provirus genome as well as the adjacent host cell integration sites. The SIP-seq droplet assay was validated using two HIV cell lines (J-Lat 15.4 and 5A8) with identical genomes but different integration sites. In comparison with shotgun sequencing, which was unable to detect any viral sequences, and nested PCR, which excluded portions of the viral genome and integration site, SIP-seq resulted in a connected contig comprising full-length viral genome and integration sites. The detection rates of the two cell lines matched the 50:50 input ratio, demonstrating no bias in recovery.
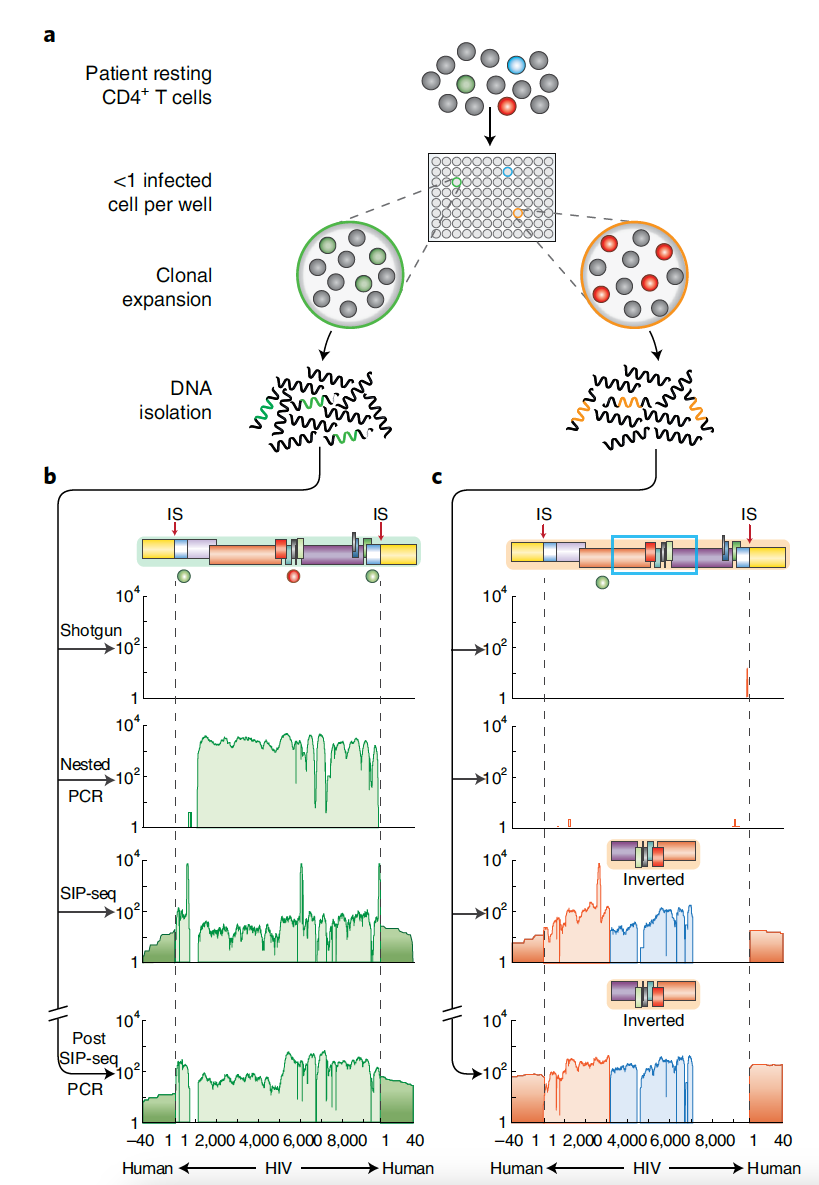
Using the same method, SIP-seq was applied to CD4+ T-cells obtained from individuals infected with HIV. First, in vitro infected cell expansion (ICE) was used to enrich the proportion of infected cells. Even in doing so, shotgun sequencing was unable to recover the HIV genome. Both nested PCR and SIP-seq were able to recover the virus genome, but again, only SIP-seq provided coverage of the host integration sites. Of note, when a second ICE culture was examined, only SIP-seq was able to identify the integrated proviral genome, suggesting that this method is very well-suited for characterizing the full genetic diversity of HIV cellular reservoirs. To test this further, SIP-seq was used to sequence DNA fragments from CD4+ T-cells taken directly from infected individuals, without ICE expansion. Of the proviral genomes recovered by SIP-seq, over 90% would not be captured by nested PCR. This demonstrates the utility of SIP-seq as a comprehensive method to study the extent of the latent HIV reservoir in ART-treated individuals. This high-throughput method is fast, cost-efficient, and easily scalable, presenting the opportunity to drastically increase the number of full-length viral genomes that can be recovered through simultaneous sequencing of multiple patient samples. Consequently, SIP-seq represents a significant advancement that will enhance our understanding of HIV persistence in cellular reservoirs.