


Droplet-based single-cell RNA-seq (scRNA-seq) is a widely adopted technique with high-throughput capabilities, essential for studying biological heterogeneity at single-cell resolution in fields like developmental biology, immunology, cancer research, and neuroscience. However, a persistent challenge in droplet-based scRNA-seq is ambient RNA contamination, which originates from nucleic acids released by dead or dying cells during single-cell processing. These ambient RNA molecules can co-encapsulate with cells in droplets or occupy empty droplets, obscuring biological signals, degrading data quality, and hindering downstream analysis. Existing post-hoc computational methods for addressing ambient RNA contamination rely on assumptions that may not universally apply, raising uncertainty about their reliability in enhancing low-quality data in all cases. Given the widespread use of scRNA-seq, the availability of tools to assess data quality is crucial for ensuring dataset comparability and reproducibility, as well as facilitating biological interpretation. This study introduces a set of metrics, available in a package called AmbiQuant, designed to evaluate droplet-based scRNA-seq data quality by quantifying ambient RNA contamination before any data filtering takes place. This study also identifies factors that contribute to ambient RNA contamination, offering experimental insights to address this issue proactively.
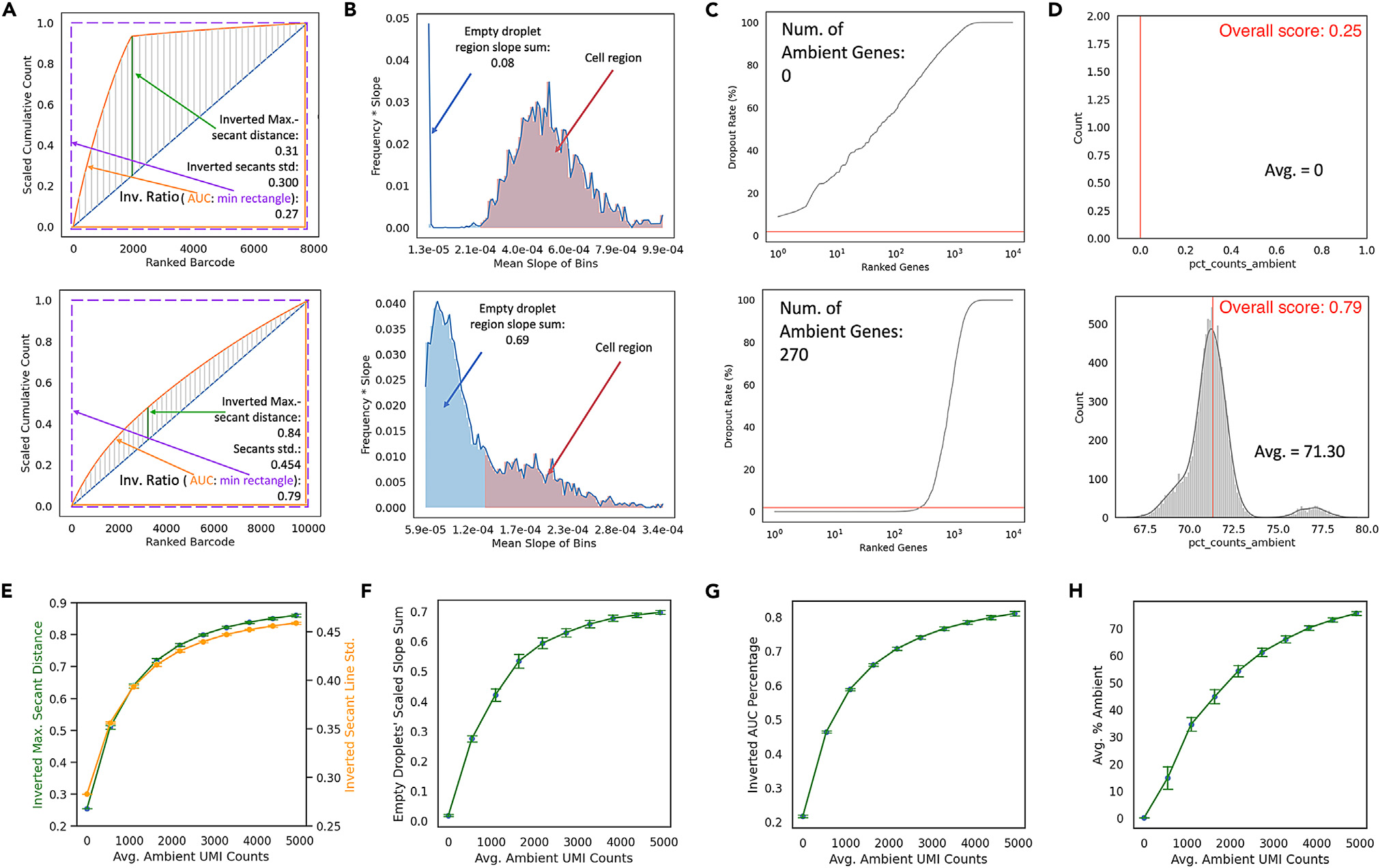
The authors aimed to create quantitative metrics for assessing ambient RNA contamination in scRNA-seq datasets prior to data filtering. They used CellBender to generate simulated datasets representing low (ambient UMI count = 100) and high (ambient UMI count = 4000) RNA contamination, while keeping other parameters, such as number of cells, number of droplets, and UMI per cell, constant. To quantify contamination, they employed geometric and statistical approaches. In the geometric approach, they analyzed the cumulative distribution of gene counts against ranked barcodes. High-quality datasets exhibited distinct slope changes, while low-quality ones had less pronounced changes due to ambient genes. In the statistical approach, they examined slope distributions in the cumulative count curve. Contaminated datasets had unimodal slope distributions, while high-quality ones showed multimodal patterns. They established a cutoff to distinguish "empty droplet" from "cell" slope distributions and quantified data points below this threshold. The contamination metrics they devised, including inverted maximal secant distance, inverted secant line standard deviation, inverted AUC percentage, sum of weighted slopes under the threshold, average percentage of ambient genes, and the number of ambient genes, all increased proportionally with the level of ambient contamination. These metrics were tested on real datasets with varying sequencing depths and contamination levels and proved robust in assessing contamination. For simplicity, they introduced an overall score on a scale from 0 (best quality, perfect signal-to-noise ratio) to 1 (worst quality, all noise), which combines multiple contamination metrics. This score provides a continuous assessment of scRNA-seq data quality based on ambient RNA contamination.
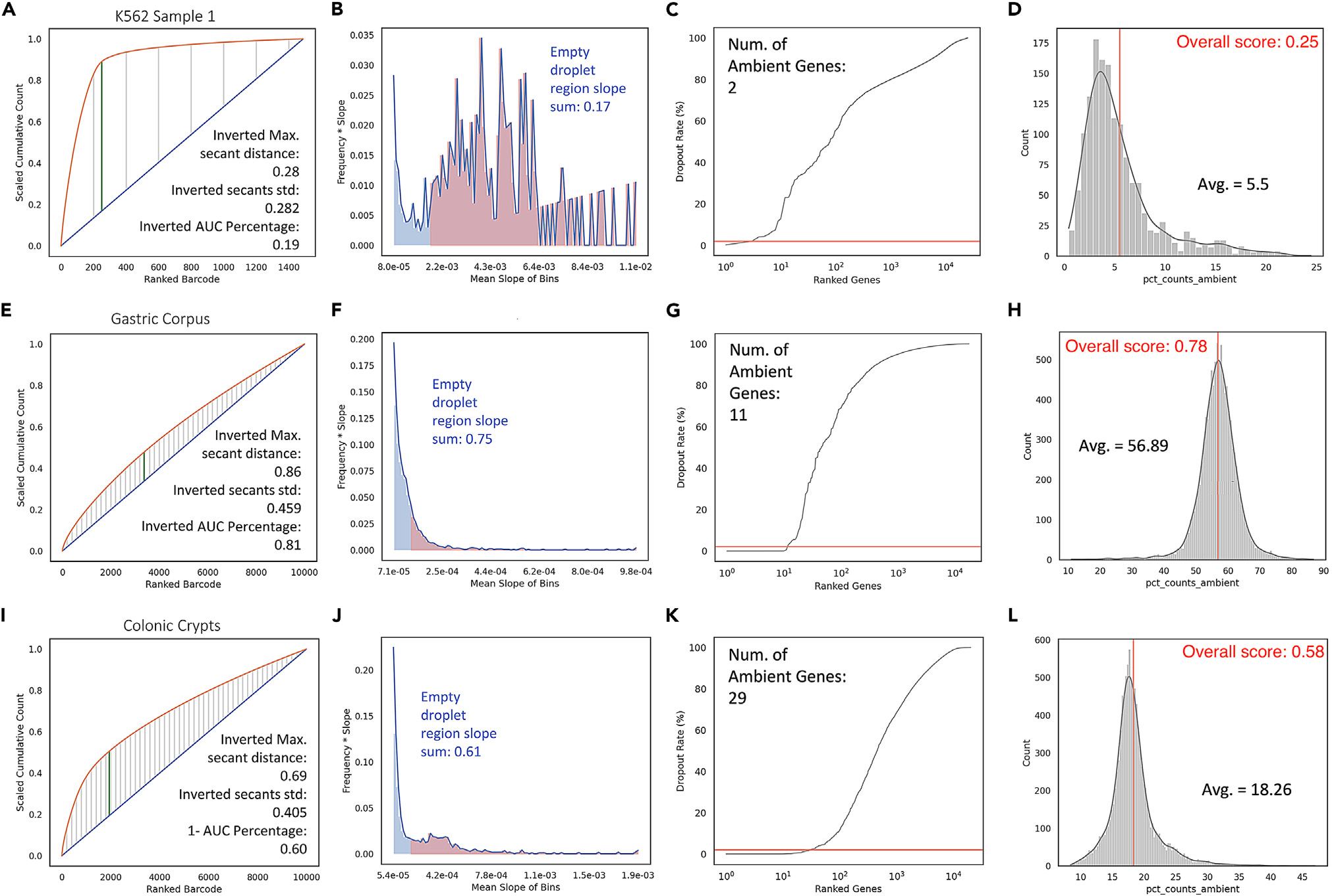
The study compared these newly developed contamination metrics with conventional quality control (QC) measures used in single-cell RNA sequencing (scRNA-seq), including total cell count, average mitochondrial gene expression, total transcripts per cell, and total genes per cell. Using the inDrops protocol, scRNA-seq datasets were generated for K562 cells, gastric corpus tissues, and colonic epithelium. The inDrops protocol has been optimized for K562 cells, and so these served as a reference for low contamination and high data quality. In contrast, gastric corpus tissues, which was dissociated in an unoptimized fashion, clearly displayed QC problems and substantial contamination. The colonic epithelium exhibited an ambient contamination issue that eluded traditional QC metrics but was detectable using the new contamination metrics. These findings emphasize the efficacy of the contamination metrics in identifying ambient RNA contamination in datasets that initially passed standard QC and underscores the significance of accounting for contamination when evaluating scRNA-seq data quality.
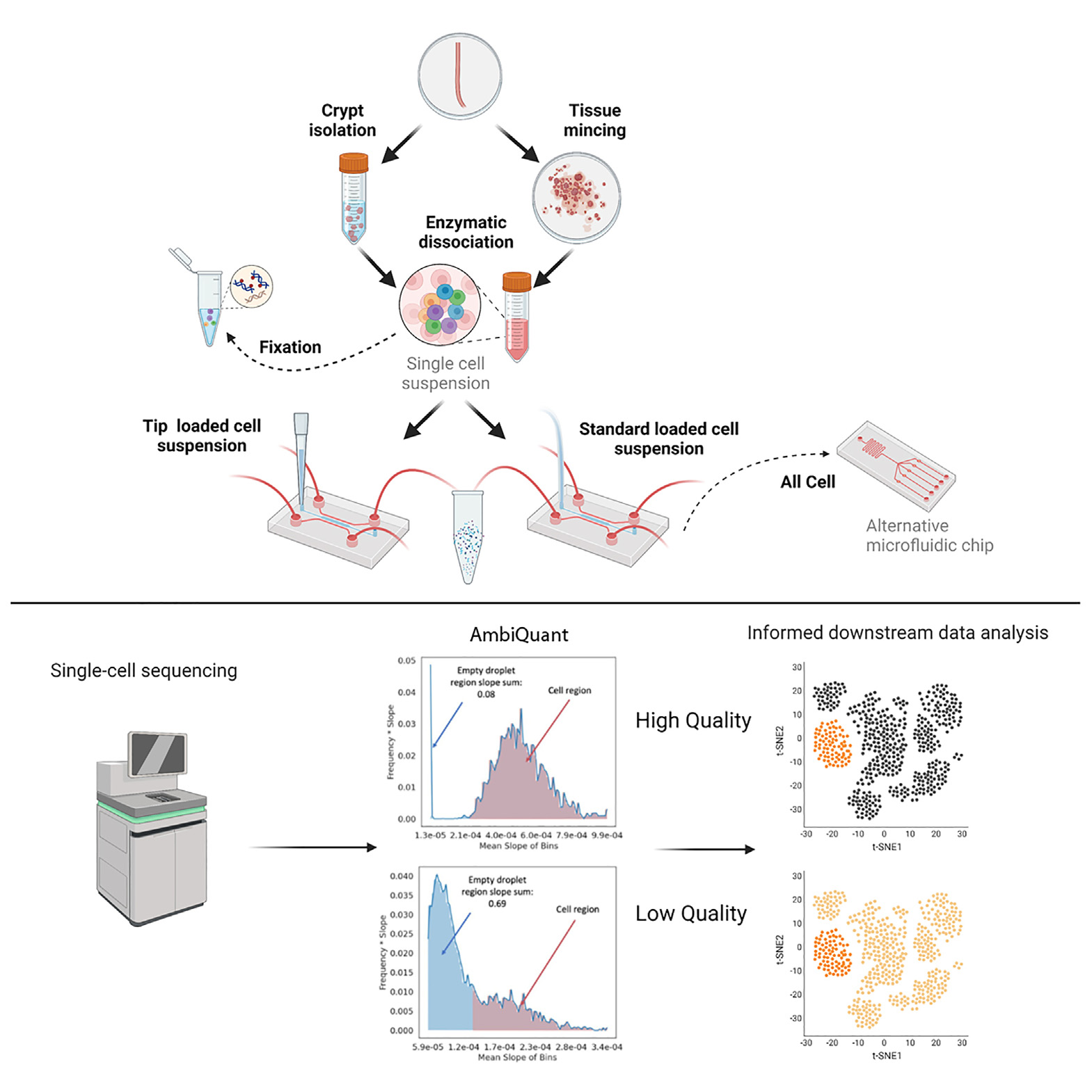
The authors next assessed how pre-encapsulation variables affect the quality of scRNA-seq data, focusing on the colonic epithelium as a model system. Using both their new contamination metrics and traditional QC measures, they examined how various tissue dissociation protocols and microfluidic manipulations impacted ambient RNA contamination. The HTAPP protocol led to cell death and damage, resulting in poor data quality as reflected by both traditional QC metrics and contamination metrics. Cold protease dissociation on isolated crypts improved data quality, reflected in lower contamination metrics and higher traditional QC metrics. Fixation immediately after dissociation also reduced ambient contamination by containing RNA within cells. They also discovered that the transversal of cells through narrow microfluidic tubing may contribute to ambient RNA contamination, but that this could be remedied by using a custom cell loading setup (tip loading). The modification also improved the data quality on colonic samples prepared with minced tissue, indicating that microfluidic manipulations can impact data quality pre- and post-encapsulation. A comparison between single-cell and single-nucleus analysis revealed that single-nucleus samples had lower data quality, as indicated by higher contamination metrics. Some techniques, such as single-cell Liberase and DNase (LD) and CD45+ depletion (CD45n), consistently yielded higher-quality data, while other protocols, such as single-cell C4 (Collagenase 4 and DNase I), led to more contaminated datasets. Differences in buffers used in single-nucleus sequencing approaches also influenced data quality. Overall, these findings highlight the significance of proper protocol selection in scRNA-seq experiments and demonstrate the utility of the AmbiQuant contamination-focused metrics in assessing data quality.
- Arceneaux D, Chen Z, Simmons AJ, Heiser CN, Southard-Smith AN, Brenan MJ, Yang Y, Chen B, Xu Y, Choi E, Campbell JD, Liu Q, Lau KS. A contamination focused approach for optimizing the single-cell RNA-seq experiment. iScience. 2023 Jun 29;26(7):107242. doi: 10.1016/j.isci.2023.107242. PMID: 37496679; PMCID: PMC10366499.