


Long-read sequencing technologies have ushered in a new era of genetic discoveries by enabling a deeper exploration into the intricate architecture of genomes. To date, however, this technology has not been employed to comprehensively characterize the entire range of genetic variations at the single-cell level, primarily due to the absence of suitable methodologies. In a recent paper for Nature Communications, Hård et al employ a droplet-based multiple displacement amplification (dMDA) reaction followed by long-read sequencing to conduct a whole genome analysis of single human cells. By bridging the gap between long-read sequencing technology and single-cell genomics, this methodology holds the potential to provide unprecedented insights into the full spectrum of genetic variation within individual cells and lays the foundation for numerous applications in both basic science and clinical research.
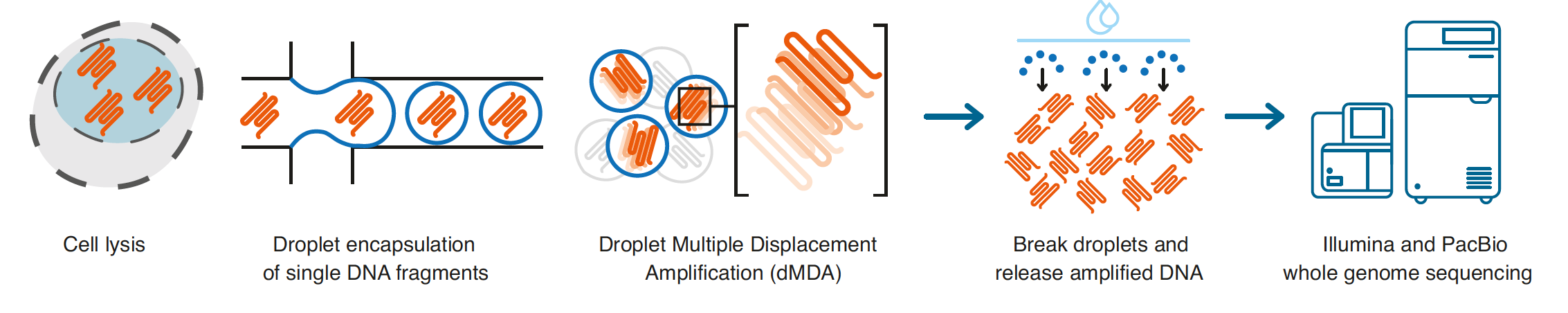
Single-cell genome sequencing is inherently difficult due to the limited number of DNA molecules present in each cell. This intrinsic constraint makes the process susceptible to allelic dropout, where one allele may be preferentially amplified over the other leading to an incomplete or inaccurate representation of the cell's genomic content. Long-read sequencing protocols also demand orders of magnitude more DNA than what is naturally present in a single human cell, necessitating substantial DNA amplification. However, the application of whole-genome amplification (WGA) can introduce undesirable effects, including amplification bias, the formation of chimeric molecules, and the risk of further exacerbating allelic dropout, compounding the challenges in single-cell long-read sequencing. The method proposed by Hård et al mitigates these challenges by using MDA, which has the capacity to amplify kb-length molecules suitable for long-read sequencing, to amplify single-cell DNA fragments within microfluidic droplets. Compartmentalization within droplets ensures that the MDA occurs in a confined volume with limited reagents and only one or a few DNA fragments. This prevents molecules from undergoing excessive over-amplification and reduces the risk of forming inter-molecular chimeras. Thus, dMDA can enhance the accuracy and reliability of the amplification process when compared to other WGA techniques.
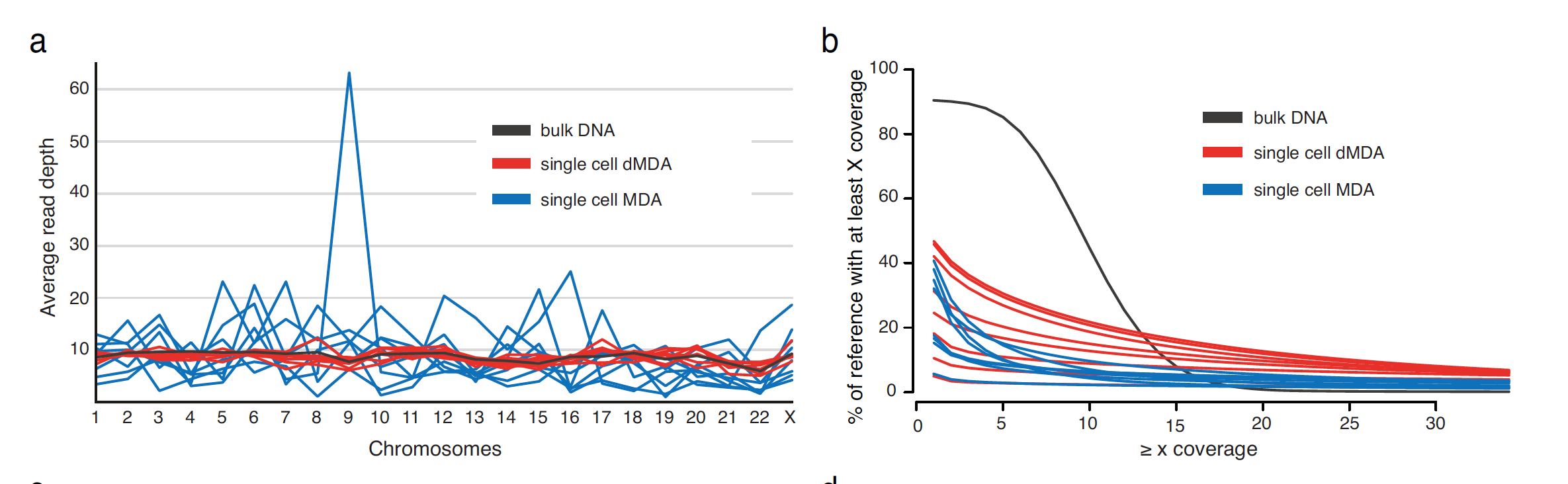
The researchers obtained two individual CD8+ T cells from the same human donor and expanded them in vitro. Fluorescence activated cell sorting (FACS) was used to isolate single cells from the culture, and these single cells were placed into a well containing lysis buffer to release the DNA fragments. The lysis buffer did not contain a fragmenting agent and 1,4-dithiothreitol was included to inhibit enzymes that fragment DNA. The resulting DNA molecules were then encapsulated in droplets along with an MDA amplification mixture that included polymerase, primers, dNTP, and reaction buffer. After incubation at 30°C for 16 h, the droplets were heat-inactivated at 65°C for 10 min and then cooled to 4°C. Droplets were then broken and the aqueous phase was collected for library preparation and whole-genome sequencing using both short-read and long-read technologies. Sixteen single-cell DNA samples (8 MDA and 8 dMDA) were initially sequenced using short-read technology. The dMDA samples demonstrated improved sequencing coverage uniformity, with fewer extreme coverage regions and a higher percentage of bases covered, consistent with prior droplet-based MDA evaluations. Five dMDA samples were then sequenced using long-read technology, achieving up to 40% genome coverage per cell.
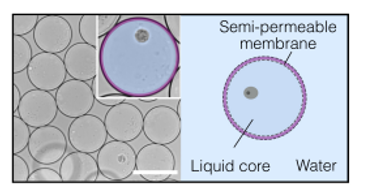
Long-read sequencing offered robust genetic variant analysis, revealing 28 somatic SNVs, mitochondrial heteroplasmy, and 5473 high-confidence structural variants. It enabled the accurate sizing of 4770 tandem repeats and the de novo assembly of two single T-cell genomes (corresponding to 15-19% of the human reference; 12.8% of genes). However, allelic dropout remained a challenge, possibly due to losses during sample preparation, droplet reagent loading, and DNA encapsulation. A promising solution could involve replacing water-in-oil droplets with semi-permeable capsules (SPCs), composed of an aqueous core enveloped by a hydrogel shell, for sample encapsulation. SPCs function like a size-selective membrane, enabling the free exchange of small reaction components while maintaining compartmentalization of single cells or large biomolecules. Thus, single cells encapsulated within SPCs could be cultured to increase available DNA. In addition, SPCs remain stable during harsh lysis conditions and permit successive washes to be used; thus, cell lysis could occur inside the compartments, rather than in well plates, and all DNA molecules would be retained. With a diameter of ~80 µm, SPCs offer efficient compartmentalization like water-in-oil droplets, enhancing the subsequent MDA reaction. Continued method refinements and the creation of specialized bioinformatics tools for single-cell long-read WGS data will advance the goal of achieving near-complete genome assemblies and full haplotype reconstructions from individual cells.