


Seasonal influenza is a major public health threat worldwide due to frequent antigenic variations that lead to the emergence of novel and potentially fatal flu strains. The segmented RNA genome of influenza A viruses (IAVs) enables viral evolution through genetic assortment during coinfections with multiple strains of IAVs. Genetic reassortment can occur across IAVs from different hosts. However, only 400-700 reassorted genomes can be genotyped in a single experiment with current sequencing or high-resolution melt analysis. In this study, the authors developed a high-throughput droplet microfluidic system to encapsulate IAV-infected cells, enabling the sequencing of 18,422 viral genomes resulting from coinfections with two circulating human IAV strains, among which 9,438 displayed reassortant genotypes. Results were highly reproducible across technical and biological replicates, confirming that genetic reassortment is nonrandom. Overall, the results demonstrate that a large proportion of reassortant genotypes can emerge upon coinfection and be detected over a wide range of frequencies using the droplet microfluidic system, which could facilitate systematic and exhaustive monitoring of the reassortment potential of IAVs for vaccine development.
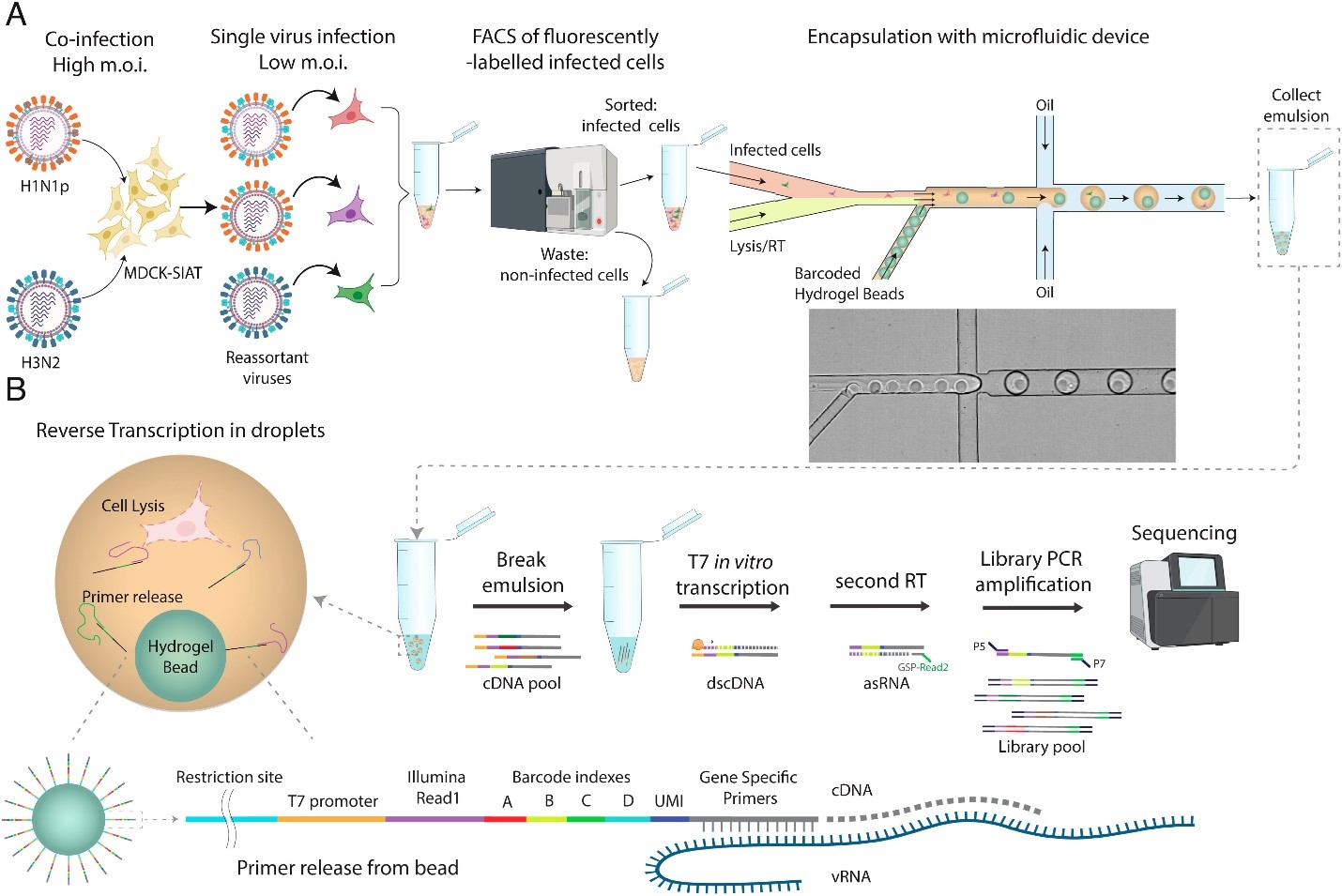
To analyze genetic reassortment during coinfections, the authors infected MDCK-SIAT cells with two human IAV strains H1N1pdm09 and H3N2 at high multiplicity of infection (MOI). Approximately a third of MDCK-SIAT cells were successfully coinfected based on IHC staining. Viral progeny from coinfected MDCK-SIAT cells, which includes reassorted genomes, was collected and then used to infect fresh MDCK-SIAT cells at low MOI. Single infected cells were then enriched by labeling cells with anti-HA and anti-M2 antibodies and collecting positive cells with fluorescence-activated cell sorting (FACS). Infected cells were then encapsulated in hydrogels with barcoded single-stranded DNA gene-specific primers for in-drop cDNA synthesis of viral genomes. The emulsions were broken to pool cDNA and a second round of transcription and cDNA synthesis was performed to reduce bias in reads between IAV segments. After excluding potential doublets and cells that did not have all eight viral segments, the authors sequenced approximately 18,000 viral genomes across 5,000 single cells.
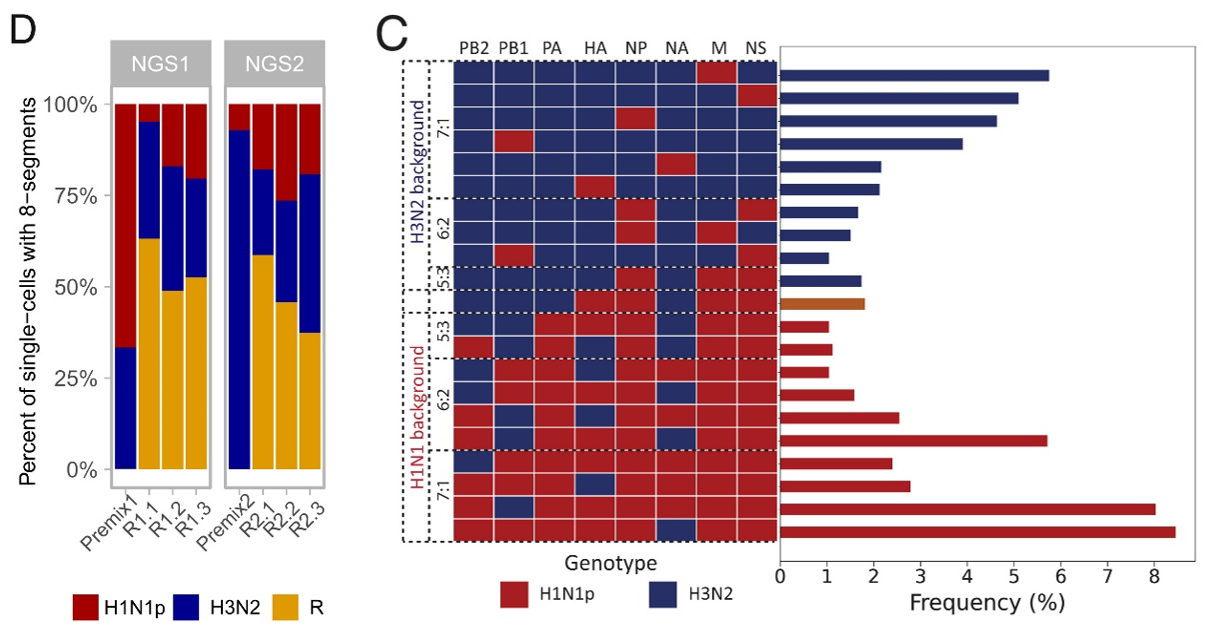
The average rate of reassortment between the two IAV strains was 51% with less than 1% of reassortants being false positives. Additionally, the reassortment rates were consistent across all replicates and thus were merged to form a single dataset. The frequencies of genotypes, single segments, and pairs of cosegregating segments were then analyzed from the merged dataset containing 2,592 reassortant eight-segment genomes. Results indicated that reassortment was non-random and that more than 60% of all possible gene combinations were generated. The distribution of genes in the reassortant genomes was nonsymmetrical, with some segments being more frequently incorporated than others. The 21 most frequent genotypes represented only 13% of detected genotypes but accounted for 66% of all reassortant genomes. Among reassortants with an H1N1p genetic background, the most frequent genotypes were 7:1 reassortants containing H3N2-NA or -PB1, or 6:2 reassortants containing H3N2-NA and -PB1.
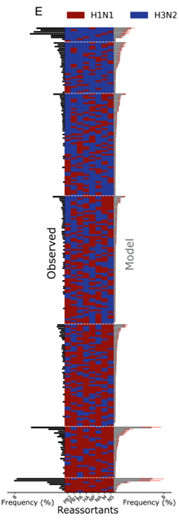
The authors next used the merged dataset to train a direct coupling analysis (DCA) model, a machine-learning technique that studies the probabilities of high-order distributions from pairwise frequencies. The researchers used a DCA model to learn parameters associated with single segments and segment pairs by maximizing the likelihood of the observed distribution of all segment combinations. The method enabled the estimation of reassortant genotype frequencies consistent with the observed eight-segment genotypes. However, the model also predicted the frequency of genotypes not observed in the eight-segment data. Notably, all genotypes were predicted to occur with non-zero frequencies, with the lowest being 2.95x10-5. The absence of certain reassortant genotypes in the experimental data from the eight-segment genome seemed to be due to the sample size. Nevertheless, this model has the potential to aid in vaccine development and identify reassortment between zoonotic animal viruses and human seasonal viruses that pose a threat to human health. The authors suggest that combining experimental data generated from the droplet microfluidic system with machine-learning methods and data mining can enable a better understanding of IAV reassortment. Overall, the work demonstrates that droplet microfluidics can be a powerful tool in studying viral evolution, with many applications beyond influenza viruses.